Opinion Article | Open Access | Published 13th October 2021
Recommendations of scientifically sound and appropriate sampling plans for parenteral drug products.
Ian Aled Jones*, Alex Bird and Nathaniel Lochrie | Cite this article | Download this PDF Paper
Abstract
This paper gives recommendations for defining sampling plans/sizes that are statistically justified or based on published guidance for typical parenteral drug products
Simple tables based on the ANSI/ASQ Z1.4 acceptance sample plans or other published guidance have been collated to aid organisations in selection of appropriate sample sizes/plans for routine drug product manufacture.
Key Words: USP <1790>, visual inspection, AQL, power, sample size
Conflict of Interest Statement
The views expressed in this paper are those of the authors and do not necessarily reflect
the policies or views of Teva UK Ltd.
1. Introduction
The current good manufacturing practice (cGMP) regulations for finished pharmaceuticals require development of controls that include scientifically sound and appropriate sampling plans. According to the revised Chapter 6 of EU GMP Guide, the sampling plan used should be appropriately justified and based on a risk management approach. Representative samples should be taken and recorded in accordance with approved written procedures.
The FDA also requires¹, that sampling should be done upon statistical criteria. However, no direct guidance is given as to the statistical criteria that should be applied by the regulatory authorities. There are numerous examples of observations and warning letters with deficiencies identified with the statistical rationale for the sampling plan presented by the organisation. This paper will summarise an approach based the use of the ANSI/ASQ Z1.4 standard and other regulatory justification.
The regulatory landscape around visual inspection does give some guidance as to sampling plans in the US pharmacopeia², which gives guidance in suggesting that typical sampling plans used for this purpose can be found in the ANSI/ASQ Z1.4 standard³, which describes a method of attribute sampling and is the most common standard used for inspection by attributes. An attribute can be defined as a characteristic or feature that is measured for each observation (record) and can vary from one observation to another, in the case of visual inspection the requirement is that the secondary inspection sampling plan, has a sufficient number of samples to represent the whole batch.
Sampling or inspection by attributes involves classifying the sampled unit as either conforming or nonconforming. It is indexed by the acceptable quality level (AQL), which is defined as “the maximum percent nonconforming (or the maximum number of nonconformities per hundred units) that, for purposes of sampling inspection, can be considered satisfactory as a process average”. The AQL isn’t lot or batch specific but rather a process average. The standard selects the sample size as a function of the population size. For any attribute sampling plan, an operating characteristic curve (OC curve) can be created that gives the probability of accepting a lot with a given defective percent in the population. The OC curve is determined by the sample size and the number of defectives allowed in the sample.
A proposed rationale to sampling for quantitative measurements is also proposed. Quantitative data are measures of values and are expressed as numbers. A quantitative test would be one where the test or assay is representative of the bulk batch after homogeneity has been proven as part of the product/process validation. For example, pH measured from a single vial may be expected to represent the pH of the total batch (if homogeneity had been proven), in reality however it would be more likely a number of vials were tested and an average value calculated. The number of samples averaged to obtain a single value would then be based on the assay method validation or the variability of the assay.
2. Attribute Sampling - Non Destructive Testing
Visual inspection for example, is a non-destructive test, the regulatory requirement is that 100% inspection is performed on each unit and after a 100% inspection using a validated inspection method, a statistically based sample should be re-inspected. For visual inspection, USP<1790>² gives guidance for batch release with a range of batch sizes and suggests sampling plans listed as Normal II are used. From this plan, the sample number with the selection of an acceptable quality limit (AQL) can be determined. Furthermore, USP<1790>² gives guidance that the accept number (the number of defective units allowed in the sample) for a critical defect is zero. Major and minor defects, which pose less risk to the patient, will have increasing (less stringent) AQL values and accept numbers greater than zero. Table 1 shows the range of AQL values suggested in <1790> for the visual inspection processes.
Table 1. Typical AQL Values for Visual Inspection Processes

Further clarity of the visual inspection process was developed by the Steering Committee of the ECA Visual Inspection Working Group⁴, which stated as per <1790> that for critical defects the accept number (the number of defective units allowed in the sample) for a critical defect was zero, rather than stating an AQL percentage. For release of the batch for major and minor defects, the minimum AQL range was defined, and is shown in Table 2.
Table 2 : Release criteria as defined in ECA Good Practice Paper “Visual Inspection of Medicinal Products for Parenteral Use (2015)”

Therefore, combining Tables 1 and 2 to give clarity on AQL % range and acceptance criteria would give the following suggested guidance.
Table 3 : Suggested guidance for Release criteria for non-destructive testing

Unfortunately, the guidance can sometimes cause confusion in quality units that are not familiar with the methodology for the ANSI/ASQ Z1.4 standard, with setting of the lowest AQL number having the unintended consequence of selecting the highest sample number defined in ANSI/ASQ Z1.4 table, irrespective of the potential batch size. It is therefore proposed that Table 4 is used which gives the lot size, the required sample number and the minimum accept number for the defined category per batch/lot size. As the accept number is always zero for critical defects, the AQL% will decrease, as the AQL% is fixed for Major and Minor defects the acceptance number will increase. It should be noted that for Lot sizes less than 1200 an assessment will need to be undertaken as to whether the AQL% is acceptable or that an increased sample size is required.
Table 4 : Acceptance number as a function of the lot size with acceptance number by defect category applying the USP guidance values for AQL% for non destructive testing Normal II.

*For lot or batch sizes less than 1200 it is suggested a risk assessment is undertaken either to justify a higher AQL level of greater than 0.1, if a sample size <125 is used.
From this table, the sample size for the “AQL” re-inspection number can be easily determined from the original size of the inspected lot and the maximum acceptance number of minor, major and critical defects are defined with the corresponding AQL %. It is for the individual organisation to determine if the maximum acceptance level defined in USP<1790> is appropriate for their process. For example a lot size of 35,000 would require a sample size of 315, with an acceptance criteria of 0 for critical defects. If an organisation made to decision to set the acceptance criteria of 0 for major defects for this lot size, then the AQL% for major defects would then reduce from 0.65% (with an accept number of 5) to 0.04% 9with an accept number of 0) for this particular category.
3. Attribute Sampling Destructive Testing
For destructive testing of attributes, where the test is of the individual unit rather than the bulk properties of the batch, for example a dissolution study on lyophilised vials, a statistically based sample size should be determined that is representative of the batch or lot tested. As the test is individual to the unit this again is described as an attribute test. Examples of these types of test would be re-constitution studies, dissolution and reconstitution time, Uniformity of Fill and Container/Closure Integrity.
Mathonet5 suggested that because QC testing, such as the reconstitution of vials is destructive to the sample, it is impractical to use AQL General Level II inspection number sampling plans due to loss of product and therefore a different approach is required. From this work, it is generally accepted that following the ANSI/ASQ Z1.4 standard3, Special Sampling Plans (AQL=0.65%) or Equivalent may be used.
Therefore using Special Sampling plan S-3, the following table was derived. It should be noted that for Lot sizes less than 3200 an assessment will again need to be undertaken as to whether the AQL% is acceptable or that an increased sample size is required.
Combining this guidance⁵ with table 2 would give the following suggested guidance.
Table 5 : Suggested guidance for Release criteria for destructive testing

Table 6 : Acceptance number as a function of the lot size with acceptance number by defect category for destructive testing

* For lot or batch sizes less than 3200 it is suggested a risk assessment is undertaken either to justify a higher AQL level of greater than 0.65%, if a sample size <20 is used.
From this table, the sample size required for attribute testing per batch/lot can be easily determined from the original size of the inspected lot and the maximum acceptance number of minor, major and critical defects are defined with the corresponding AQL %. Again, it is for the individual organisation to determine if the maximum acceptance level defined is appropriate for their process.
4. Other Regulatory Expectation Attribute Sampling
There are specific sampling requirements that have defined guidance for attribute sampling of Sterility, Endotoxin and Sub-visible particles.
4.1 Attribute Sampling - Sterility
Sterility testing of finished product is performed to provide assurance of the sterility of products in order to ensure that they are safe for human use. Sterility testing should be performed in accordance with US Pharmacopoeia (USP) section <71>, European Pharmacopoeia (Ph. Eur) Section 2.6.1 and Japanese Pharmacopoeia (JP) Section 4.06.
A portion of the batch is sampled based on the overall batch size and product type in line with USP <71> Table 3 (Minimum Number of Articles to be Tested in Relation to the Number of Articles in the Batch). Table 3 defines the minimum number of items to be tested for each medium. Two different test media, typically Soybean-Casein Digest Medium (TSB) and Fluid Thioglycollate Medium (FTM), are utilised therefore requiring 20 containers per medium for Parenteral preparations with a batch size >500 units. A further rationale for sterility sampling is also defined⁶ in 21 CFR Ch. I (4–1–12 Edition) which states: “The sample used for each test medium or each incubation temperature of a test medium for the final container and first repeat final container test shall be no less than 20 final containers from each filling of each lot, selected to represent all stages of filling from the bulk vessel.” Two different test media (TSB and FTM) are utilised giving therefore >20 vials per medium.
4.2 Attribute Sampling - Endotoxin
Endotoxin testing of raw materials, intermediate and finished product is performed in accordance with USP <85>, Ph. Eur section 2.6.14, and JP section 4.01. However, sampling plan information is addressed in AAMI ST72, for describing methods and calculation of pyrogen and endotoxin testing. Sampling plan information is addressed in⁷ AAMI ST72, which recommends that for routine production endotoxin testing; for batch sizes >101, that the number of samples analysed should be 3% of the lot, up to a maximum of 10. The FDA recommends that finished product samples for analysis of bacterial endotoxin can be pooled into a composite sample. Pooled extracts can be kept at stability temperatures until tested. Therefore, the batch may be tested with vials sampled start, middle and end and pooled into 3 test samples.
4.3 Attribute Sampling –Sub-Visible Particles
USP-NF 〈788〉⁸ states that for small-volume parenterals less than 25 mL in volume, the contents of 10 or more units are combined in a cleaned container to obtain a volume of NLT 25 mL; the test solution may be prepared by mixing the contents of a suitable number of vials and diluting to 25 mL with particle-free water.
5. Quantitative Sampling
Where the test or assay is representative of the bulk batch and homogeneity has been proven as part of the product/process validation, then the sample size that is required for a quantitative assay is either determined from the assay method development validation report or from a calculation of power for the assay.
When an assay is to measure a quantitative characteristic (for example a compositional characteristic measured on a continuous scale such as pH) where the batch has been shown to be homogenous, it is imperative that the assay is powered adequately to ensure that the assay result truly represents the batch. The minimum power required for any study is considered to be 80%⁹. A “power” of 0.8 (80%) indicates that there is an 80% chance of demonstrating equivalence when the difference (or ratio) between the population means is actually within the equivalence limits. The higher the statistical power for a given assay, the lower the probability of making a false negative error. That is the higher the probability of detecting an effect when there is an effect. Sample size is positively related to power, increasing the sample size and averaging the value will increase the power of the assay.
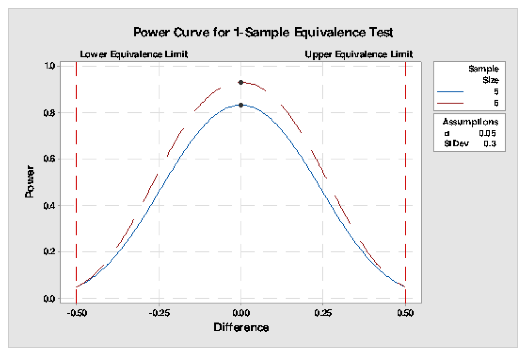
An example of a calculation of power for an assay is shown, assuming a pH assay has a target of 5, with limits of +/-0.5 and a standard deviation of the assay determined as 0.3, then the number of samples required to be averaged to obtain a sufficient power for the assay may be calculated by the use of statistical software such as Minitab®. For this example, a power of 83% required a sample size of 5 and power of 93% required a sample size of 6 using a 1 sample equivalence test with α =0.05. The power curve for this example is shown in Fig 1.
Fig 1 : Power curve for a single sample equivalence test for an assay with acceptance range of =/-0.5 with a standard deviation of the assay of 0.3.
6. Summary
Table 7 summarises the justification for sampling sizes/plans for typical parenteral drug products, with recommendation for statistically based sample sizes for AQL re-inspection, QC and microbiological testing.
Table 7: Summary of samples sizes/plans based upon statistical criteria or guidance


Conclusion
When sampling plans are required to be established for parenteral drug products, the use of the tables suggested, may be a simpler approach for personnel that are not expert with the use of the ANSI/ASQ Z1.4 standard. The summary (table 7) gives guidance to the approach in terms of defining sampling plans/sizes that are statistically justified or based on published guidance.
The paper gives the minimum requirement for sample sizes, it is for the individual organisation to determine if a greater sample size is required to minimise the AQL% and to also determine when smaller batches/lots (>3200) are tested that the sample size selected is appropriate against the AQL% or that the sample size is increased.
References
01. FDA Code of Federal Regulations (21 CFR Part 211.84)
02. USP:41(6) In-Process revision: <1790> Visual Inspection of Injections
03. ANSI/ASQ Z1.4-2003 (R2018): Sampling Procedures and Tables for Inspection by Attributes
04. ECA Good Practice Paper “Visual Inspection of Medicinal Products for Parenteral Use (2015)
05. PDA J Pharm Sci and Tech 2016, 70 392-408 (A Biopharmaceutical Industry Perspective on the Control of Visible Particles in Biotechnology-Derived Injectable Drug Products)
06. FDA Code of Federal Regulations (21 CFR Ch. I (4–1–12 Edition))
07. ANSI/AAMI ST72:2019 Bacterial Endotoxins - Test Methods, Routine Monitoring, And Alternatives To Batch Testing
08. USP-NF 〈788〉 Particulate Matter in Injections
09. Basic Statistics and Pharmaceutical Statistical Applications by James E. De Muth Pg 621 CRC Press and Hintze JL. Kaysville, Utah, USA: 2008.
Author Information
Ian Aled Jones*, Alex Bird and Nathaniel Lochrie
*Corresponding Author aled.jones@tevaruncorn.co.uk
Affiliations:
Teva UK Ltd, Whitehouse Vale Aston Lane North, Runcorn WA7 3FA
Kommentare